
Garbage in, garbage out: A critical evaluation of strategiesused for validation of immunohistochemical biomarkers
Authors: Gillian O’Hurley, Evelina Sjostedt, Arman Rahman, Bo Lia,
Caroline Kampf, Fredrik Ponten, William M. Gallagher,
Cecilia Lindskog
Online: Free online access
Issue: Mol Oncol. 2014 Jun;8(4):783-98.
PMID: 24725481
Abstract
The use of immunohistochemistry (IHC) in clinical cohorts is of paramount importance in
determining the utility of a biomarker in clinical practice. A major bottleneck in translating
a biomarker from bench-to-bedside is the lack of well characterized, specific antibodies
suitable for IHC. Despite the widespread use of IHC as a biomarker validation tool, no universally
accepted standardization guidelines have been developed to determine the applicability
of particular antibodies for IHC prior to its use. In this review, we discuss the
technical challenges faced by the use of immunohistochemical biomarkers and rigorously
explore classical and emerging antibody validation technologies. Based on our review of
these technologies, we provide strict criteria for the pragmatic validation of antibodies
for use in immunohistochemical assays.
1. Introduction
The classical method of immunohistochemistry (IHC) allows
for visualization of specific antigens in tissues or cells
based on antibody-antigen recognition, using brightfield or
fluorescent microscopy. The history of IHC goes back to the
early 1940s, when Coons and colleagues detected antigens in
frozen tissue sections by developing an immunofluorescence
technique (Coons et al., 1941). Introduction of a method based
on peroxidase-labelled antibodies opened the door to development
of more advanced approaches (Mason et al., 1969;
Nakane, 1968), enabling IHC to be used on routinely processed
tissue sections, such as formalin-fixed paraffin-embedded
(FFPE) tissues. However, it took until the early 1990s for the
method to become generally accepted in diagnostic pathology
(Leong, 1992; Taylor, 1994).
IHC is today a widely used method that can be rapidly performed
in most laboratories. The procedure is short, simple
and cost-effective. Indeed, IHC has emerged as an important
tool to detect cellular markers defining specific phenotypes
relative to disease status and biology. Moreover, IHC is utilized
for basic and clinical research, from small projects to highthroughput
strategies, to evaluate potential biomarkers in
clinical patient cohorts. However, the lack of standardized
guidelines for determining the specificity and functionality
of antibodies renders the translation of promising biomarkers
to the clinic difficult. Herein, we discuss the various limitations
and technical challenges that need to be addressed
when using IHC for biomarker development and clinical
validation.
2. Review of clinically used IHC markers approved
by FDA
A biomarker is defined as a molecule that is objectively
measured and evaluated as an indicator of normal biological
process, pathogenic process, or pharmacological responses to
therapeutic intervention (Biomarkers-Definitions-Working-
Group, 2001). Although great efforts have been made in the
last decade to discover novel cancer biomarkers for use in
clinical practice, a striking number of these efforts fail to
make it into the clinic (Fuzery et al., 2013). One of the causes
of this failure of translation could be the limited knowledge
that scientists working in biomarker discovery have in
analytical, diagnostic and regulatory requirements for clinical
assays (Fuzery et al., 2013). Over the last few decades a
number of key FDA approved cancer biomarkers have been
introduced into the clinic for differential diagnosis of specific
tumours, leading to improvement of cancer detection and
staging, identification of tumour subclasses, prediction of outcome after treatment, and selection of patients for different treatment options. However, of these approved biomarkers,
only five are individual IHC-based biomarkers
(Fuzery et al., 2013) (Table 1). The earliest FDA approved biomarkers
for IHC application were assays to detect the estrogen
receptor (ER), progesterone receptor (PR) and HER-2/neu
(c-erbB-2). The presence of these biomarkers in breast cancer
tissue serves as a diagnostic, prognostic and predictive
method to assist pathologists in identifying breast cancer
subtypes and determine whether patients are suitable candidates
to receive certain targeted therapies such as Tamoxifen
(ER positive patients) or Trastuzumab (Her-2 positive patients).
The IHC biomarker c-kit (CD117), which is used in
the clinic to detect gastrointestinal stromal tumours (GISTs)
(Debiec-Rychter et al., 2004), and p63, which is used to detect
the presence of basal cells indicative of normal prostate
glands (Shah et al., 2002; Weinstein et al., 2002), are the latest
FDA approved single marker IHC-based assays which were
approved almost a decade ago in 2004 and 2005, respectively.
Since then no other individual biomarker developed for
detection in an IHC assay has been FDA approved. However,
despite lack of FDA approval, there are many IHC markers utilized
in some clinics to assist pathologists in diagnosis and
decision making. Such examples include the use of E-Cadherin
and/or p120 staining to assist diagnosis of invasive
lobular breast carcinoma (Rakha et al., 2010), various antibody
panels for diagnosis and sub-classification of malignant
lymphomas, as well as the use of the proliferating nuclear
marker, Ki67.
An ideal biomarker demonstrating clinical sensitivity and
specificity of 100% is almost never achieved in practice due
the fact that increasing one of the parameters is only
achieved at the expense of the other. As a result, panel
biomarker assays are becoming more relevant. Two emerging
IHC panel-based assays are Mammostrat by Clarient InsightDx and IHC4 by Genoptix Medical Laboratory. Mammostrat is an IHC-based panel assay that can estimate risk
of recurrence in hormone receptor-positive, early stage
breast cancer patients which is independent of proliferation
and grade. This assay quantifies p53, HTF9C, CEACAM5,
NDRG1 and SLC7A5 by a defined mathematical algorithm
resulting in a risk index (Bartlett et al., 2012, 2010). Similarly,
IHC4 is another emerging assay which estimates recurrence
risk for early stage breast cancer patients by quantifying
IHC measurement of ER, PR, HER2 and Ki-67 using Aqua
technology (Cuzick et al., 2011).
IHC-based biomarker assays represent an attractive
approach for biomarker detection in the clinic as the IHC technique
is routinely carried out in clinical laboratories, there is a
fast turn-around time from assay to results and it is costeffective.
However, the paucity of FDA-approved biomarkers
for IHC-based assays emphasizes the importance and urgent
requirement of standardized guidelines and workflows for
IHC assay development which should be implemented at an
early stage of biomarker discovery. This will ensure robust
analytical and clinical performance and ultimately lead to a
better chance of an IHC-based biomarker assay achieving
FDA approval.
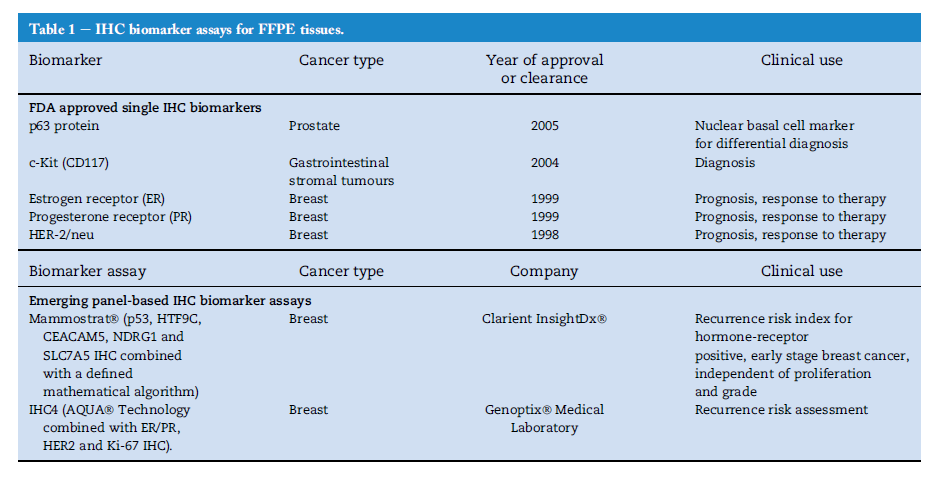
3. Review of factors influencing the IHC process
The standard brightfield IHC technique is comprised of three
components; slide preparation, IHC procedure and interpretation.
Antibodies used in the clinic have undergone thorough
testing and every step of the protocol has been well established,
including both positive and negative controls. Factors
which may affect the outcome of IHC include tissue handling,
epitope retrieval, storage and handling of tissue sections,
choice of antibody, detection method and interpretation procedure.
To yield the expected staining pattern when establishing
a new antibody, all factors which may influence the
standardization and reproducibility of the process need to be
carefully considered. These factors are summarized in
Figure 1 and will be described more in detail below.
3.1. Tissue handling immediately after surgery, fixation
and processing
‘Ischemia time’ refers to the time from when a tissue or organ
is cut off from O2 supply through removal of a specimen from
the body in surgery, to fixation of the specimen. Ischemia
results in degradation of protein, RNA and DNA, as well as
activation of tissue enzymes and autolysis (Kumar et al.,
2005) and can therefore be a major factor influencing IHC results.
Recently, Pekmezci et al. demonstrated that longer
cold ischemia time affects the detection of ER and PR by IHC
in breast cancer (Pekmezci et al., 2012). Although the American
Society of Clinical Oncology and College of American Pathologists
(ASCO/CAP) has developed guidelines for handling
of tissues for ER, PR and HER-2 detection in breast cancer patients,
such guidelines are not available for other surgical
specimens (Comanescu et al., 2012; Hammond et al., 2010).
Fixation is another critical step in the IHC process to preserve
tissue morphology and retain antigenicity of the target molecules.
Two types of fixatives are commonly used in histopathology;
(1) non-coagulating fixatives (formaldehyde,
glutaraldehyde, osmium teroxide, potassium dichromate
and acetic acid) and (2) coagulating fixatives (alcohol, zinc
salts, mercuric chloride, chromium trioxide and picric acid).
The most common fixative used in histopathology is 10%
neutral-buffered formalin. This is composed of 4% paraformaldehyde
solution which is buffered to a neutral pH.
Formalin cross-links peptides by formation of hydroxymethyl
groups on reactive amino acid side chains, providing excellent
preservation of tissue architecture; however, formalin fixation
can mask epitopes and result in decreased antigenicity.
Several factors influence the formalin fixation method, such
as temperature, time, penetration rate, specimen dimension,
volume ratio, pH of the buffer and osmolality, but unfortunately,
there is a lack of available guidelines to establish a
standard practise across pathology laboratories.
3.2. Appropriate storage and handling of tissue sections
Another factor that may influence the IHC outcome is storage
of prepared tissue sections (Wester et al., 2000; Williams
et al., 1997). It has been suggested that storing tissue sections
longer than two months leads to loss of p53 antigen reactivity
(Prioleau and Schnitt, 1995). The mechanisms underlying the
loss of antigenicity in FFPE tissue is unclear. It has been
hypothesised that oxidation may be the key contributor of
antigenicity loss (Blind et al., 2008; Sauter and Mirlacher,
2002). Due to this and the fact that degradation of protein is
temperature dependent, a large variety of storage conditions
for cut sections have been advocated such as cold storage, paraffin coating or vacuum sealed desiccators. However, recently it has been suggested by Xie et al. (2011) that the
presence of water both endogenously and exogenously plays
a central role in loss of antigenicity. Therefore, slide storage
conditions that are protected from oxidization by vacuum
storage or paraffin coating are not completely protecting
slides from loss of antigenicity if residual water from inadequate
tissue processing is present on the tissue (Xie et al.,
2011). Thus, the optimal storage of unstained sections is yet
to be defined, making freshly cut sections or sections stored
for less than two months most ideal. For long-term storage,
vacuum containers or storage in colder conditions (þ4/18)
is often recommended.
3.3. Appropriate and efficient epitope retrieval
Another major step that should be considered carefully when
performing IHC is antigen retrieval (AR). The two methods of
antigen retrieval are (1) heat-induced epitope retrieval (HIER)
(e.g. citrate pH 6.0, TriseEDTA pH 9.0 and EDTA pH 8.0) and
(2) proteolytic enzyme-induced epitope retrieval (PIER) (e.g. proteinase
K, trypsin, pepsin, pronase). Of the two methods, HIER
is most commonly used. The technique was first described by
Shi and colleagues (Shi et al., 1991) and has been improved by
a number of investigators (Cattoretti et al., 1993; Greenwell
et al., 1991, 1993) for its routine use in laboratories throughout
theworld. However, the mechanisms of AR are not fully understood.
It is speculated that both HIER and PIER serve to break
the methylene bridges created during fixation, exposing the
antigenic sites in order to allow the antibodies to bind
(D’Amico et al., 2009; Fowler et al., 2011; Kakimoto et al., 2008;
Leong and Leong, 2007; Suurmeijer and Boon, 1993).
There are several different AR variables that can affect IHC
staining results such as heating, the choice of AR solution, its
pH and molarity, and the effect of metal ions (D’Amico et al.,
2009; Emoto et al., 2005). An appropriately controlled AR
method can restore antigenicity in formalin fixed paraffin
embedded (FFPE) tissue to resemble the antigenicity of frozen
tissue. Moreover, it can facilitate IHC standardization, despite
variations in tissue fixation and subsequent handling (von
Boguslawsky, 1994) (Shi et al., 2007; Taylor, 2006). However,
the appropriate AR protocol is dependent on both the antibody
and the target protein, and needs to be optimized for every antibody.
3.4. Appropriate choice of antibody (monoclonal vs
polyclonal)
The three cardinal points that must be considered when
buying commercial primary antibodies for IHC are as follows:
(1) use reliable, recommended companies, (2) obtain complete
information about the antibody to ensure it is applicable or
recommended for IHC and, (3) characterize the specificity of
the antibody. A significant number of commercial antibodies
are not thoroughly analysed for off-target binding, e.g. using
protein arrays (Chang, 1983; Nilsson et al., 2005). In addition,
several companies do not provide the sequence of the antigen
the antibody was raised against (Saper, 2009) and, therefore,
antibody validation is a mandatory step before proceeding
with IHC.
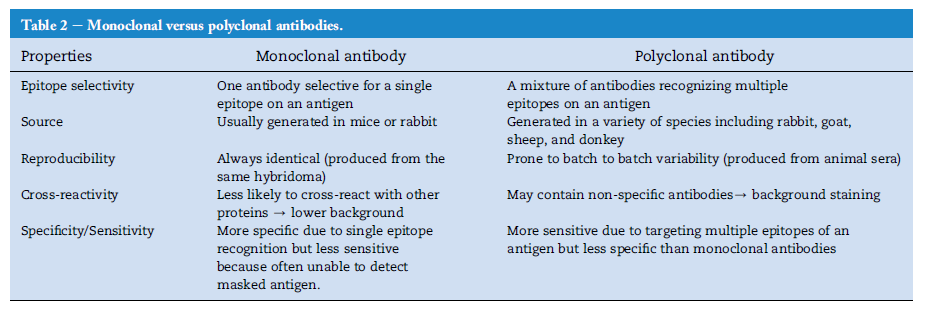
The choice of using either monoclonal or polyclonal primary
antibodies for IHC further complicates the issue of
epitope specificity and determining which antibody would
be more suitable for IHC (Bordeaux et al., 2003). Polyclonal
antibodies are a collection of antibodies targeted against
multiple epitopes of a particular antigen. Generally, when
an animal is injected with a specific antigen, the immune
system elicits a primary immune response by producing
multiple B cell clones against the antigen. After subsequent
immunization with the same antigen, these B cells differentiate
into plasma cells producing and secreting antibodies
found in the serum. The serum containing polyclonal antibodies
can be affinity purified using the antigen as a ligand,
which eliminates 99% of antibodies recognizing other targets
than the antigen. This procedure results in antibodies
with higher specificity than conventional polyclonal antibodies,
still retaining the ability to recognize different epitopes
on the same antigen (Lindskog et al., 2005). A
monoclonal antibody is generated by selection of one single
B cell from spleen or bone marrow of the immunized animal
and fusing this cell with immortal myeloma cells to
produce hybridoma cells (Kohler and Milstein, 1975). As
such, the culture supernatant contains only one type of
antibody specific for a single epitope of the immunizing
peptide. The advantages and disadvantages of using polyclonal
and monoclonal antibodies for IHC are summarized
in Table 2.
A useful tool to search for appropriate antibodies suitable
for IHC is the portal, Antibodypedia (http://www.antibodypedia.
com). Here, antibodies are listed with reference to antibody
companies and associated validation data (Bjorling and
Uhlen, 2008).
3.5. Use of a sensitive and robust detection system
The outcome of an IHC assay depends on the use of sensitive
protein detection system in order to visualize the antigeneantibody
reaction. The most popular methods of detection
are enzyme and fluorophore-mediated detection
systems. With chromogenic substrates, an enzyme label is
reacted with the substrate to yield a strong colour product
visualized by brightfield imaging. Alkaline phosphatase (AP)
and horseradish peroxidase (HRP) are the two most extensively
used enzymes, both with available chromogenic, fluorogenic
and chemiluminescent substrates.
Detection systems in IHC can be divided into two broad categories,
namely direct or indirect. In the direct detection
method, the primary antibody is labelled with enzymes or
fluorochromes, enabling direct detection of the antigen on
the tissue section without the requirement of a secondary
antibody. This method of detection is simpler and less time
consuming; however, it has the disadvantage of lower sensitivity
compared with indirect methods. The indirect detection
method involves the use of unlabelled primary antibodies and
labelled secondary/tertiary antibodies, which are specific for
the bound primary antibody. Although this method is time
consuming and complicated by multiple steps, indirect detection
method is more sensitive in detecting tissue antigens.
Some commonly used indirect detections mechanisms are
as follows; the avidin-biotin complex (ABC) method, the
labelled streptavidin biotin (LSAB) method, the phosphataseanti-
phosphatase (PAP) and the polymer-based detection
system. There are several other immunohistochemical detection
methods such as tyramide amplification, cycled tyramide
amplification, fluorescyl-tyramide amplification and rolling
circle amplification, but these are not heavily used to date in
routine IHC.
3.6. Detection of phosphorylation using IHC
Post-translational modifications are important biological
events that control the behaviour of a protein. Phosphorylation
is a post-translational process regulating protein activity
by the addition and removal of a phosphate group.
Tissue phosphoproteomic studies show promise for the
discovery of key phosphorylated proteins and signalling
pathways in many diseases (Bodo and Hsi, 2011). The
detection and quantification of phosphorylation has been
well established using techniques such as Western blotting
on cell lysates but it represents a new era in diagnostic pathology.
Many phospho-specific antibodies have been
generated for immunohistochemical application; however,
the detection step remains challenging due to the labile
nature of phosphorylated proteins, reflecting dynamic processes.
In addition, tissues become oxygen deficient
shortly after being isolated from the blood supply and subsequently
undergo rapid protein dephosphorylation (Blow,
2007). Therefore, if the tissues are not fixed within 60 min
post-surgical removal from the living body, the majority of
phospho-epitopes are lost (Baker et al., 2005; Jones et al.,
2008). Due to this, most phosphorylation studies have not
been reproduced. Other variations between studies leading
to these discrepant results can include sample procurement,
processing, scoring/quantification and subjectively
selected cut-offs (Bodo and Hsi, 2011). Therefore, rigorous
standardization of laboratory procedures for tissue preservation
and for the overall IHC technique as well as quantification
is required for success in quantifying
phosphorylation by IHC in tissue. Post-translational modifications
such as phosphorylation can also be studied with
proximity ligation assay (PLA), described in Section 4.3.
However, many of the same issues discussed here will
also apply to PLA.
3.7. Use of manual immunohistochemistry versus
automated immunohistochemistry platforms
A major milestone in the standardization, reliability and
reproducibility of IHC is the invention of automated IHC platforms.
Many critical steps in the manual IHC method are
operator-dependent and essential to the quality of the final
IHC result and its reproducibility (Shi and Taylor, 2011). These
include the critical antigen retrieval step, reagent preparation,
application of reagents, appropriate washing steps and multiple
incubation times. The use of automated IHC not only allows
for larger volumes of slides to be stained
simultaneously under standardized conditions, but also provides
assistance to operators through additional processing
monitoring errors such as alarms for inappropriate temperatures,
insufficient volumes of reagent, expired reagents and
even the selection of an incorrect reagent via the use of barcode
scanning (Fetsch and Abati, 1999; Moreau et al., 1998;
Prichard et al., 2011).
Many automated IHC machines, particularly those used in
a clinical setting, are what is termed as “closed systems”
which means the instrument is closed to introducing variations.
Although this is an important advantage for standardization
of IHC staining, it can be a drawback for research as the
flexibility of choosing reagents, retrieval methods and introduction
of subtle variation to the technique is lost. This has
led to the development of “open” automated systems, offering
similar flexibility as manual staining (Prichard et al., 2011).
However, as HIER is not performed on an “open” platform,
some of the same limitations of manual IHC discussed previously
apply to this type of automated IHC. Clearly, there are
advantages and disadvantages to the manual staining method
and the “open” and “closed” automated systems so the choice
of method should be influenced by the laboratory’s purpose
(Prichard et al., 2011). However, for large-scale IHC efforts
where planning and standardized IHC protocols are necessary
(Uhlen et al., 2005; Warford et al., 2004) it can be anticipated
that automated IHC may lead to reduction in error rate as
each step of the staining procedure is recorded (Howat et al.,
2014). Together with tissue microarray (TMA) technology
(Battifora, 1986; Kononen et al., 1998), where a large number
of tissues from different organs or individuals are assembled
on a single slide, high-throughput IHC minimizes reproducibility
issues.
3.8. Interpretation via manual and automated
approaches
Manual assessment of IHC staining remains the traditional
method for most diagnostic and predictive decisions in pathology.
However, manual interpretation of IHC data can be
time intensive, laborious and an inherently subjective and
semi-quantitative process (Fiore et al., 2012). Observer variability
can exist in three forms; intra-observer variability,
inter-observer variability and inter-laboratory variability
(Conway et al., 2008). The latter is usually attributed to issues
regarding tissue fixation and processing, antibodies used and
detection systems. Intra-observer variability, referring to the
lack of consistent assessment by the observer, occurs less
frequently than inter-observer variability due to the fact that
pathologists adhere to their own internal standards (Kay
et al., 1994). Inter-observer variability is the greatest problem
associated with human-based assessment of IHC staining,
influenced by factors such as misplaced orientation on a
TMA slide, eye fatigue, complexity of data management
following differential categorical scoring, quality of microscope,
illumination of microscope and individual human
vision limitations (Conway et al., 2008).
Utilizing image analysis systems on virtual microscopy
slides or whole slide images has been proposed as solving
the problem of standardized quantification of IHC data, due
to its capability of producing continuous datasets eliminating
categorical and biased assessment. High-throughput image
analysis methods can also reduce workloads and outperform
human manual scoring in terms of reproducibility and precision,
as they are not affected by fatigue or subjectivity. Enormous
advances in image analysis systems on tissue sections
have been achieved over the years (Taylor and Levenson,
2006). However, despite these advances, image analysis is
far from ready to replace the expert pathologist, as it is still
very much a semi-automated approach as most algorithms
require specific input and training by a pathologist in order
to produce accurate output. In addition, image analysis approaches
are highly influenced by a number of factors that
can affect the quality of their performance. For example, the
quality of sections/TMAs hugely affects the resulting data obtained
from image analysis. This is due to the inability of most
of the current automated image analysis systems to identify
irregularities on a section that the human eye can ignore,
such as artefacts, edge effect staining, folding of tissue and
thickness of tissue section, which may produce a false score.
Moreover, image analysis often fails to distinguish tumour
from benign tissue. Nevertheless, it is widely accepted that
the continuous development of computer-aided image analysis
technologies will lead to quantitative systems that will
compliment and support the pathologist/human expert to
produce a less subjective and accurate IHC assessment.
3.9. Multiplexing: brightfield vs. darkfield
When measuring protein expression levels in tissue, a decision
must be made as to whether assessment should be performed
by IHC using brightfield imaging or
immunofluorescence (IF) using fluorescent imaging, where
both techniques offer advantages over the other. Brightfield
imaging utilizes visible white light to illuminate the tissue,
and protein expression is classically observed and graded
based on the intensity of 3,30-diaminobenzidine (DAB), generating
a brown staining (Gustashaw et al., 2010). Counterstaining
with haematoxylin keeps morphological detail of the
surrounding tissue intact and allows visualisation and analysis
of localized protein. The IF technique visualizes protein
expression in tissue against a dark background, using an antibody
with a chemically attached fluorochrome, such as fluorescein
isothiocyanate (FITC) or tetramethyl rhodamine
isothiocyanate (TRITC) (Jordan et al., 2002). The antigeneantibody
complex can be visualized using a fluorescent imaging
instrument such as a microscope or scanner.
IHC using brightfield imaging is one of the pillars of modern
pathology and a fundamental research tool in both
pathology and translational research (Robertson and Savage,
2008), due to the many advantages associated with the technique.
It can be performed routinely on FFPE tissue, which
permits a pathologist or researcher to work with a familiar,
conventional microscope (Jordan et al., 2002). In addition, it
can detect antigens expressed at relatively low levels due to
chromogenic enhancement steps, the equipment cost is low,
and only minimal laboratory space is required. Most importantly
in a clinical setting, the chromogens are very stable
and long-term slide storage is possible for many years. However,
as a research tool, there are some major limitations associated
with the technique. Firstly, the resolution of antigen
localization is limited due to the chromogenic substrate precipitate,
as well as the thickness of the sections imaged in
the light microscope. Secondly, saturation of chromogenic
systems occurs easily, which restricts quantitative analyses
(Robertson and Savage, 2008). Above all, IHC using brightfield
microscopy has a narrow dynamic range limiting its capability
of multiplexing, and as cross reactivity is common, three antibodies/
chromogens at a time is a maximum. Therefore,
sequential or multi-step staining is crucial to ensure
cross reactivity does not occur with enzymes used or with
primary/secondary antibodies raised in the same species.
In addition, choosing colour combinations that are
distinguishable by eye from each other and from the counterstain
can be challenging, particularly when looking at colocalized
proteins. The concentration of precipitate may also
inhibit further reaction, making it difficult to visualize rare
targets and highly abundant targets on the same slide
(Christensen and Winthers, 2009). Moreover, quantitation of
multi-staining using brightfield microscopy is even more
limited, as most brightfield image analysis tools are primarily
designed to quantify single chromogens. However, the use of
spectral imaging technologies allows unmixing of stains and
individual quantification of each chromogen.
In contrast to brightfield IHC, IF has a better capability of
multiple labelling, as IF is of higher resolution due to the fluorophores
being directly conjugated to the antibody (Robertson
and Savage, 2008). Although choosing dyes with distinguishable
spectral properties is still an issue, fluorescent imaging
has a much broader dynamic range compared with brightfield
imaging (Christensen and Winthers, 2009). On the other hand,
IF-based detection presents certain difficulties in respect to
interpretation of tissue morphology, as well as the cost of reagents
and equipment. Moreover, a fluorescent signal can be
quenched when the fluorophores are in close proximity, and
as fluorophores are not as stable as chromogens, photobleaching
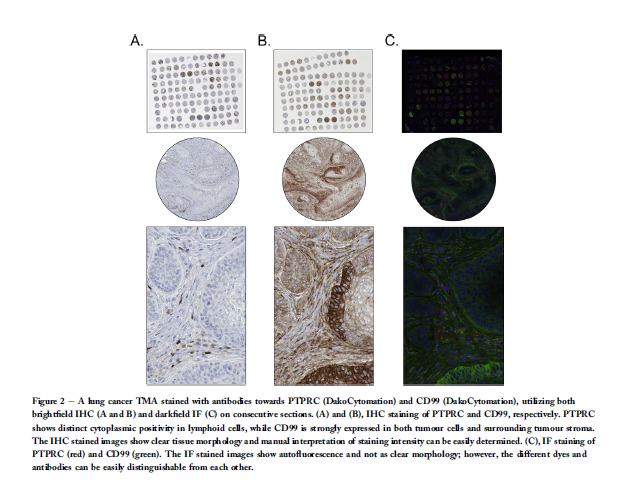
of stored slides is an issue. The most restraining aspect of IF is inherent autofluorescence of FFPE material, making high quality immunofluorescence imaging capricious (Robertson
and Savage, 2008) and limiting the use of clinical material. Examples
of consecutive sections stained with both brightfield
and darkfield are displayed in Figure 2, illustrating some of
the advantages and disadvantages with both methods.
The use of multispectral imaging has overcome many of
the issues regarding autofluorescence on FFPE tissue
(Mansfield et al., 2005; Robertson and Savage, 2008). However,
many reports using IF labelling of FFPE sections (Bataille et al.,
2006; Bossard et al., 2006; Ferri et al., 1997; Hoover et al., 1998;
Mason et al., 2000; Niki et al., 2004; Nurnberger et al., 2006;
Papaxoinis et al., 2007; Scott et al., 2004; Suetterlin et al.,
2004) have not been widely acknowledged by the scientific
community (Robertson and Savage, 2008) rendering IHC by
brightfield microscopy a more accepted assay for clinical use
in quantifying protein expression. However, continuous
research and development of new methods in the area of IF
and image analysis, such as the new technique MxIF (Gerdes
et al., 2013), will bridge the gap between classical IHC of FFPE
material and the acceptance of IF analysis of human FFPE
tissues.
One potentially might also consider application of both
brightfield and fluorescent imaging, e.g. use of H&E staining/
brightfield imaging for localisation of tumour regions and
use of fluorescence-based imaging for quantitation of consecutive
tissue sections.
4. Review of currently used validation methods for
antibodies for IHC
Commercial production of antibodies is well established;
however, there are no universally accepted guidelines or standardized
methods for determining the validity of these reagents
(Bordeaux et al., 2003). The production and validation
of specific antibodies is a challenging, costly and time
consuming process. Perhaps as a result, the quality control
by the antibody vendors is not always what it should be
(Couchman, 2009). Moreover, the information supplied in academic
publications where the antibodies are used is often
insufficient. Therefore, it is imperative that investigators
take requisite steps to assure themselves that the specificity
of each antibody is as advertised. Here we explore both classical
and emerging technologies for antibody validation.
4.1. Which staining pattern is expected?
The signal intensity is generally related to the antibody concentration
(Dabbs, 2006). In order to get an optimal dilution
of an antibody, rendering the greatest contrast between
desired (specific) positivity and unwanted (non-specific) background,
it is necessary to know which staining pattern to
expect. Hence, the first crucial step in antibody validation is
to understand the nature of the target protein. For wellknown
or partly characterized proteins, information
regarding the expected staining pattern can be obtained
from available databases such as Uniprot (www.uniprot.org),
the Human Protein Atlas (www.proteinatlas.org), or by
searches in published literature. Bioinformatic prediction
algorithms for expected subcellular localisation, including
presence of signal peptides or transmembrane regions, is
gathered in online sources such as MDM (Fagerberg et al.,
2010), SPOCTOPUS (Viklund et al., 2008) and Phobius (Kall
et al., 2004). Furthermore, information on post-translational
modifications or splice variants is important in order to
predict detection of multiple bands in Western blotting.
Such information can be retrieved from e.g. OMIM
org). A large fraction of the human proteins are essentially
uncharacterized and experimental data is needed for validation
of the generated staining pattern in IHC.
4.2. Western blotting
The standard antibody validation method is Western blotting,
whereby antibody specificity is confirmed by the presence of a
single band corresponding to the predicted molecular weight
of the target protein. However, as many proteins have a
similar molecular weight, a band of the correct size is not
full evidence for targeting the intended protein. Moreover,
the kinetics of antibody-antigen binding is context dependent
and validation needs to be performed in an applicationspecific
manner. Therefore, even if an antibody yields a
band of correct predicted size in Western blotting, it does
not necessarily imply that the antibody is functional in IHC assays
on FFPE tissue. This is mainly due to the fact that immunogenic
epitopes are exposed differently in SDS-PAGE
compared to formalin fixation. Proteins are denatured during
the Western blotting process so post-translational modifications
on the native protein may not be represented, while
epitope masking (Hawkes et al., 1982) can occur with formalin
fixation. Furthermore, as Western blot is dependent on the
relative concentration of both the target and other proteins
in the sample, even antibodies validated as highly specific
may generate cross-reactivity to off-target proteins in the
sample. This may be overcome by using cell lysates overexpressing
the full-length target protein, as the probability of
correct protein detection is higher when a protein is present
at sufficiently high level (Algenas et al., 2014).
4.3. Paired antibodies and proximity ligation assay
Paired antibodies are defined as antibodies raised against
different, non-overlapping epitopes on the same target protein.
A similar IHC staining pattern yielded by two separate
antibodies towards the same target protein on consecutive
sections suggests a higher level of reliability, especially of
importance for proteins lacking previous characterization
(Uhlen et al., 2010). A dissimilar staining pattern does not
however necessarily imply that both antibodies are unspecific,
as one of them still could show the correct pattern. In
addition, dissimilar antibodies could potentially mean that
the antibodies are directed towards different isoforms of the
same target protein, and other methods are necessary to
decide if the antibody is specific. Even a similar staining patterns
obtained by a set of paired antibodies can be difficult
to interpret, and do not conclude if the two antibodies display
the same unspecific background. The latter can be further
elucidated using in situ proximity ligation assay (PLA).
The PLA technique is highly sensitive method determining
protein interactions and analysing post-translational modifications
(Blokzijl et al., 2010; Lizardi et al., 1998; Soderberg
et al., 2006). It is based on the principle that two or more
oligonucleotide-conjugated antibodies need to bind in close
proximity in order to detect a signal, and can be utilized
directly in frozen or FFPE tissue sections (Soderberg et al.,
2008; Zieba et al., 2010). The binding is visualized by labelling
the oligonucleotides with fluorophores or HRP. As two separate
binding events are required to produce a signal, PLA
also serves as a useful and reliable tool for antibody validation,
using antibodies directed towards different epitopes on the
same target protein. The signal generated by PLA can be quantified,
and as each event produces a single “dot”, the outcome
can be measured more easily compared to IHC staining intensity,
facilitating automated image analysis.
4.4. Comparison with RNA sequencing data
The central dogma suggests a direct relationship between
mRNA expression and protein levels in a population of cells
at steady state. Lately, development of RNA sequencing
(RNA-Seq) has provided sensitive and reproducible expression
analyses which can be easily applied for large scale exploration
(Brawand et al., 2011; Wang et al., 2009). Comparison
with transcription data may be a valuable antibody validation
tool, whereby the quantitative measurement of the transcript
abundance can be used to support the validation of protein
expression. Several comprehensive RNA expression datasets
are available online, e.g. at the Human Protein Atlas
(www.proteinatlas.org) (Fagerberg et al., 2013), the RNA-Seq
atlas (www.medicalgenomics.org) (Krupp et al., 2012) and
the BioGPS portal (www.biogps.org) (Wuet al., 2009). However,
expression and abundance data is more noisy and complex
than the underlying genomic sequence information, and protein
levels are influenced by translational and posttranslational
mechanisms. Some proteins are secreted or
transported to other sites, and may not be observed in the organ
where mRNA is expressed. This is the case for e.g. liver,
where a large set of genes displaying high liver-specific
mRNA expression are negative for the corresponding proteins
in liver, while positive in plasma (Kampf et al., 2014). Hence,
some proteins may be present at levels not readily predicted
by mRNA levels (Ghaemmaghami et al., 2003;
Schwanhausser et al., 2011). On the contrary, a high correlation
between mRNA and protein levels has still been shown
in a number of studies (Greenbaum et al., 2002; Lu et al.,
2007). The molecular pathways determining the expression
patterns need to be further elucidated, in order to answer
the fundamental question to what extent mRNA and protein
expression correlate.
4.5. In situ hybridization
The RNA-Seq technique may provide quantitative measurements
of transcript levels; however, the comparison to IHC
data is quite crude. The sequence mRNA pool from a tissue
sample reflects all the different cell types present in the sample,
and the RNA-Seq lacks the precise localization and high
cellular resolution provided by IHC. For morphological
information on spatial distribution, in situ hybridization (ISH)
uses RNA probes labelled with e.g. biotin that can be visualized
in FFPE tissues (Carson et al., 2002; Gall and Pardue,
1969; Jin and Lloyd, 1997). One example of a large-scale initiative
using ISH spatial data is the Allen Brain Atlas (Lein et al.,
2007), extensively used in the field of neuroscience. ISH renders
a staining that can be compared with that of IHC and
may thus serve as an antibody validation technique, e.g. identifying
false positive results (Kiflemariam et al., 2012). However,
as for several other methods, blocking of endogenous
peroxidase and biotin could be a limiting factor (Qian and
Lloyd, 2003), and in addition, ISH lacks the sensitivity to distinguish
between sequences of high homology.
4.6. Mass spectrometry
Mass spectrometry provides the standard for detecting and
quantifying a targeted set of proteins in a sample. The
method uses the principle of ionizing peptides derived by
proteolysis, and measuring the signal intensity of fragment
ions over time, which indicates the abundance of the peptide
in the sample (Anderson and Hunter, 2006; Towbin et al.,
1979). As mass spectrometry yields a quantitative measurement
of the target protein, it may be an important complement
in validating the expression pattern rendered by an
antibody, i.e. in analysing unexpected bands yielded by Western
blotting. However, mass spectrometry lacks the spatial
resolution that can be provided by IHC, and has sensitivity
problems. It has been shown that the signal response of
different peptides from the same protein can vary as much
as 100-fold in intensity (Picotti et al., 2007). Mass spectrometry
also has a bias towards highly expressed proteins, as a
low detection limit results in a reduced signal-to-noise-ratio
(Hack, 2004; Lange et al., 2008).
4.7. Appropriate positive and negative cell/tissue
controls
Another approach to ensure antibody specificity is to perform
IHC on positive and negative FFPE control cell lines known to
express or not express the target protein, and to perform
Western blotting on their subsequent lysates. This is also a
useful tool to ensure your antibody is applicable to use on
FFPE material prior to its use on valuable FFPE tissue. However,
cell lines in which targets have appropriate levels of
expression or lack of expression can be limited. In these instances,
alternative approaches of cell manipulation can be
performed to create positive and negative control cells. Overexpression
models can be created and used as positive controls
by introducing viral constructs that contain the gene/
protein of interest into a cell line via lentiviral or retroviral
transduction or plasmid-based transfection (Seth, 2005). Similarly,
negative control cell lines can be derived by RNA interference
(RNAi), whereby expression of a target gene can be
knocked down with high specificity (Rao et al., 2009). Alternatively,
the use of the recently developed approach of clustered
regularly interspaced short palindromic repeats (CRISPR) (Cho
and Kim, 2013; Mali et al., 2013) could be used to generate a
negative control. Unlike RNAi knockdown where transfection
efficiency rarely reaches 100%, the CRISPR approach allows for complete knockdown which is ideal for insurance of antibody specificity. In addition, the use of tissue where a knockout of
the gene has been engineered can be used to argue specificity
of the primary antibody. It must also be noted that there is an
increasing provision of commercial recombinant cell lines on
the market with either ectopic overexpression of specific proteins
(e.g. from Origene Technologies Inc.) or knockouts in cell
lines (e.g. Horizon Discovery Ltd.).
4.8. Other commercially available controls
Many other techniques available through antibody suppliers
can be carried out on tissue to test for antibody specificity.
Isotope controls can be used to control for cross-reactivity.
This method ensures that the staining observed is not a result
of immunoglobulins binding non-specifically to Fc receptors
present on the cell surface. However, the method does not
prove that the antibody is binding to the target antigen.
Synthetic peptides towards which the commercial antibodies
were generated can be used in competitive assays,
where antiserumis incubated with the synthetic peptide prior
to staining. If the staining component of the antiserum is
raised against that antigen, the antibodies should adsorb to
the peptide and little or no staining should be observed
(Saper, 2005). However, although this is an acceptable assay
for validation of polyclonal antibodies, the technique cannot
be used for monoclonal antibodies as they will always be
adsorbed by their antigen, even if they are staining something
entirely different in the tissue (Saper, 2005). Furthermore,
even as a polyclonal antibody validation tool, it does not rule
out that other tissue proteins cross-react with the synthetic
peptide.
5. Ideal work-flow
There does not exist an unflawed antibody validation method
for IHC, and each method has its own advantages and disadvantages.
In this section, we describe and discuss two alternative
recommended work-flows to follow in order to ensure an
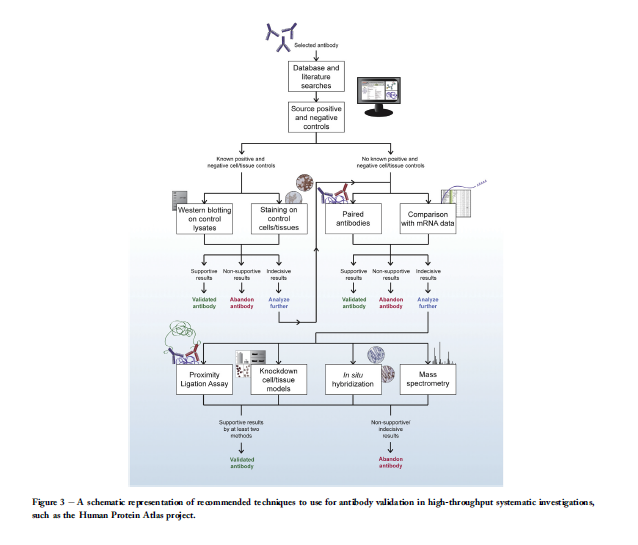
antibody is of highest quality prior to use in IHC. One is
intended for IHC in high-throughput strategies, such as the
Human Protein Atlas project (Figure 3), and one is suitable
for IHC in mainstream biomarker development applications,
particularly those intending clinical application (Figure 4).
Both approaches firstly involve the identification and selection
of an appropriate antibody, and searches of literature and
databases in order to fully understand the target protein and
identify positive and negative controls. In the case of well
characterized differentially expressed genes, IHC staining on
cell lines or tissues known to express or not express the target
protein is a relatively inexpensive, fast and an easily assessed
method. Ideally, the validation should be complemented by
Western blotting of the corresponding cell or tissue lysates.
Previous experiments suggest, however, that a large fraction
of all proteins are expressed in a house-keeping manner
(Fagerberg et al., 2013; Ponten et al., 2009). For such ubiquitously
expressed proteins, this validation strategy has
limitations as to lack of negative controls, as almost any antibody
could render a ubiquitous staining pattern in IHC
depending on the antibody concentration used. In addition,
many proteins are largely uncharacterized and a more thorough
investigation needs to be performed in order to ensure
the antibody binds to its intended target.
The recommended antibody validation techniques to
consider next largely rely on the cost and time that can be
spent for thorough validation, and the laboratory’s access to
tissues and certain equipment. Moreover, it needs to be taken
into consideration that the desired level of accuracy and specificity
versus sensitivity may differ depending on the aim of
the study. A biomarker intended to be used for labelling of
beta cells in pancreas may only require absent staining in
other cells of islet of Langerhans and abdominal organs adjacent
to pancreas, while unspecific antibody binding in other
tissues does not interfere with the result (Lindskog et al.,
2012). In contrast, a potential diagnostic marker with the
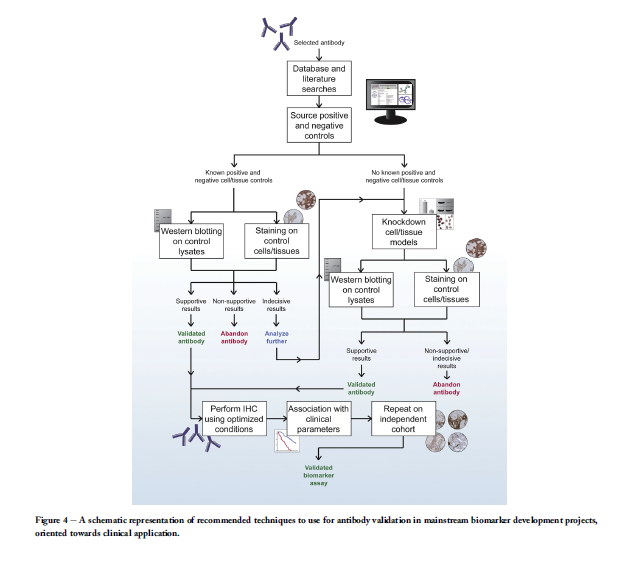
aim to accurately determine the origin of a metastasis tumour
needs a higher level of specificity, in order to set the correct
diagnosis (Gremel et al., 2014). The strategies also differ between
validation of antibodies in high-throughput projects
and antibodies intended to be used in biomarker assays.
One example of a high-throughput IHC initiative is the Human
Protein Atlas project, which systematically explores the
human proteome using in-house generated affinity purified
polyclonal antibodies on TMAs (Kampf et al., 2012; Uhlen
et al., 2005). The TMAs contain samples from 44 different
normal tissues, the 20 most common cancer types, 46 cell
lines and six samples of primary cells. The current publically
available version of the online atlas (www.proteinatlas.org)
covers 16,621 human genes, represented by data from 21,984
antibodies, and thus serves as a valuable resource in
biomarker discovery (Asplund and Edqvist, 2012; Ponten
et al., 2011). The Human Protein Atlas utilizes paired antibodies
and comparison with mRNA data, which in conjunction
with IHC staining on test TMAs and Western blotting
suggest a high level of antibody reliability. In challenging
cases where the obtained results are contradictive or indecisive,
thorough investigation with other methods such as
PLA, knockdown models, in situ hybridization or mass spectrometry
could add potential value in determining an antibody’s
specificity. A flow-chart recommended for such highthroughput
projects is displayed in Figure 3.
In oncology drug research and development, where researchers
seek to introduce drugs targeted to molecular pathways
and reduce development timelines, there is an
increasing demand for specific and sensitive cancer tissuebased
IHC biomarkers (Smith and Womack, 2014). The two
most critical elements of a successful IHC assay are reliable
antibodies and tissue sample integrity, and a failure to validate
these elements sufficiently will lead to conflicting, irreproducible
results (Smith and Womack, 2014). Therefore, we
propose a strict but appropriate IHC workflow that should be
adhered to for research and development of potential biomarkers
(Figure 4). In this workflow, inclusion of definite positive
and negative FFPE controls is imperative in every IHC run
where antibody specificity can be verified as well as controlling
for additional run variations. These controls may be in
the form of either cell or tissue controls. Moreover, the use
of automated systems is recommended to limit errors due to
technical and laboratory variability.
6. Discussion and conclusion
IHC is an invaluable validation tool in biomarker discovery.
However, considering the excessive number of existing
studies proposing novel IHC biomarkers, markers validated
in several clinical cohorts are extremely few, stressing the
need to raise quality standards for clinical biomarker studies.
Even if results can be reproduced, the transition towards a
routinely used marker is complex. For a new factor to become
of potential value in the clinic, it has to add an important value
compared with other already used factors. Moreover, it also
has to be taken into account in which patient material the factor
was analysed and if it fits with the population where it
potentially will be used. To be able to perform and reproduce
a multitude of studies for the same marker, a specific antibody
and standardized antibody validation workflow is crucial. We
agree with the proposal recently made by (Howat et al., 2014),
suggesting that the antibody conditions should be published
on an open access site following publication in order to keep
the knowledge already gained by research groups. This would
aid in protocol optimization, minimize waste of valuable patient
material and improve the quality of publications.
In this review, we described and discussed methods available
for the validation of antibodies prior to usage in IHC, as
well as numerous factors in the IHC procedure that can potentially
influence the end result. In addition, we provide strict
criteria that should be adhered for the pragmatic validation
of antibodies for use in both high-throughput, systematic investigations
and mainstream biomarker discovery-oriented
immunohistochemical assays.